{kind=link}
FDA Class I Recall: Understanding the Most Serious Medical Device Classification
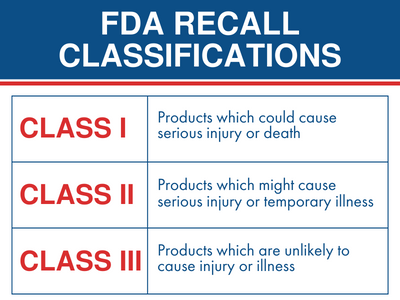
The U.S. Food and Drug Administration (FDA) issues a Class I recall when there is a reasonable probability that the use of, or exposure to, a violative product will cause serious adverse health consequences or death. This is the agency’s most urgent classification, reserved for medical devices that pose an immediate, life-threatening risk to patients.
What Defines a Class I Recall?
The FDA categorizes medical device recalls into three distinct tiers based on the level of risk the product poses to the public. According to the FDA’s official guidance, a Class I recall is the most severe level.
Unlike Class II recalls—which involve products that may cause temporary or medically reversible health problems—or Class III recalls—which involve products unlikely to cause any adverse health reaction—Class I recalls are initiated when a device is defective to the point that it could lead to permanent injury or fatal outcomes. The agency mandates that manufacturers notify both healthcare providers and patients immediately when such a device is identified in the field.
The Process of an FDA Recall

When a manufacturer or the FDA identifies a safety concern, the agency evaluates the potential health risk. If the risk meets the threshold for a Class I designation, the manufacturer must remove the product from the market or correct the defect.
Key steps in this process include:
- Public Notification: The FDA publishes the recall on its Medical Device Recalls database to ensure transparency.
- Communication: Manufacturers are required to send “Urgent Medical Device Recall” notices to all facilities or individuals currently using the product.
- Correction or Removal: Depending on the nature of the issue, the company must either retrieve the device, repair it, or provide software updates to mitigate the risk.
Why These Recalls Matter
The primary purpose of a Class I recall is to prevent patient harm. Because these devices are often used in critical care settings—such as hospitals, surgical suites, or home monitoring—the failure of these products can be catastrophic.
For example, past Class I recalls have involved cybersecurity vulnerabilities in infusion pumps or hardware defects in heart valves. By identifying these risks early, the FDA and manufacturers work to prevent incidents before they manifest in clinical environments.
Frequently Asked Questions

How do I know if a device I use is under recall?
The FDA maintains a searchable public database where you can look up specific devices by brand name or manufacturer. If you are a patient, your healthcare provider is typically the first point of contact for recall notifications.
Is a Class I recall the same as a market withdrawal?
No. A recall is a corrective action taken against a product that violates FDA law. A market withdrawal occurs when a company removes a product from the market for minor violations that do not warrant legal action, or because of a business decision rather than a safety defect.
What should I do if a device is recalled?
Do not stop using a life-sustaining device without consulting your doctor first. Contact your healthcare provider immediately to discuss alternative treatment options or to determine if your specific unit is affected by the recall notice.