{kind=link}
Familial angiolipomatosis is a rare, benign condition characterized by the development of multiple subcutaneous angiolipomas, which are fatty tumors containing prominent blood vessels. Unlike sporadic angiolipomas, this familial variant typically follows an autosomal dominant inheritance pattern. Diagnosis relies on clinical presentation and histopathological confirmation, while management focuses on surgical excision for symptomatic relief or cosmetic concerns.
Understanding Familial Angiolipomatosis
Familial angiolipomatosis is a clinical entity distinct from solitary angiolipomas due to its hereditary nature and the presentation of multiple lesions. According to the National Institutes of Health (NIH) Genetic and Rare Diseases Information Center, these tumors are most frequently located on the trunk and extremities. They typically emerge during early adulthood, often appearing after puberty.
The condition is defined by the presence of mature adipose tissue mixed with capillary proliferation. While these tumors are non-cancerous, their tendency to occur in clusters can cause significant discomfort and aesthetic distress for patients. Clinicians emphasize that while the condition is benign, it requires careful differentiation from other lipomatous conditions, such as Dercum’s disease or multiple symmetric lipomatosis, which present with different clinical features and underlying metabolic associations.
Diagnostic Criteria and Clinical Assessment
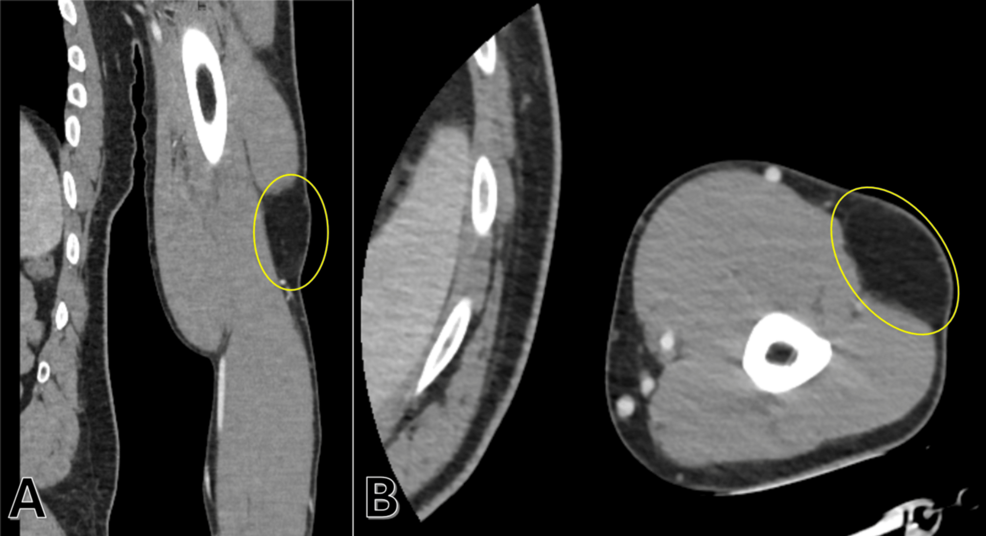
Diagnosis of familial angiolipomatosis begins with a physical examination of the subcutaneous nodules. Because these lesions are often tender, clinicians use palpation to identify the characteristic vascular components within the fatty mass.
According to research published in the journal Cureus, definitive diagnosis requires a biopsy and histopathological analysis. The presence of mature fat cells and thin-walled, branching capillaries is the hallmark of an angiolipoma. In familial cases, a detailed family history is essential. Because the condition is autosomal dominant, at least one first-degree relative often displays similar clinical findings, which helps distinguish it from isolated, non-hereditary angiolipoma cases.
Key Diagnostic Challenges

- Differential Diagnosis: Distinguishing between familial angiolipomatosis and other soft-tissue tumors like liposarcoma or angiolipoleiomyoma.
- Imaging Requirements: Ultrasound or Magnetic Resonance Imaging (MRI) is often used to map the extent of the lesions before surgical intervention.
- Genetic Counseling: Given the hereditary nature, patients are often referred for genetic consultation to understand the transmission risks for offspring.
Management and Surgical Considerations
There is no pharmacological cure for familial angiolipomatosis. Treatment is reserved for lesions that cause pain, limit movement, or result in significant cosmetic issues. Surgical excision remains the gold standard for removing symptomatic tumors.
Surgeons face unique challenges in managing these patients, particularly in underserved settings where access to specialized dermatological surgery may be limited. According to the American Academy of Dermatology, complete removal is vital to prevent recurrence, though the development of new lesions in different locations is common in familial variants. In cases where multiple tumors are present, clinicians often prioritize the excision of the most painful or rapidly growing lesions to minimize surgical trauma and recovery time.
Why Early Recognition Matters
Early identification allows for proper monitoring and prevents unnecessary anxiety regarding the nature of the growths. Because angiolipomas are benign, many patients do not require aggressive intervention if the lesions are asymptomatic. However, the familial aspect necessitates a comprehensive approach that includes tracking the patient’s family members.
Comparing familial angiolipomatosis to solitary angiolipoma highlights the importance of patient history. While a solitary angiolipoma is generally considered a localized, non-recurring event, familial angiolipomatosis requires a lifelong management plan. This distinction ensures that patients receive appropriate monitoring for new nodules rather than being treated for an isolated incident, providing a clearer path for clinical care and patient expectations.