{kind=link}
Understanding Sickle Cell Disease: Symptoms, Diagnosis, and Management
Sickle cell disease (SCD) is a group of inherited red blood cell disorders that cause red blood cells to become hard, sticky, and C-shaped rather than round and flexible. According to the Centers for Disease Control and Prevention (CDC), these misshapen cells die early, leading to a shortage of healthy red blood cells—a condition known as anemia—and frequently blocking blood flow, which causes severe pain and organ damage.
What Are the Common Symptoms of Sickle Cell Disease?
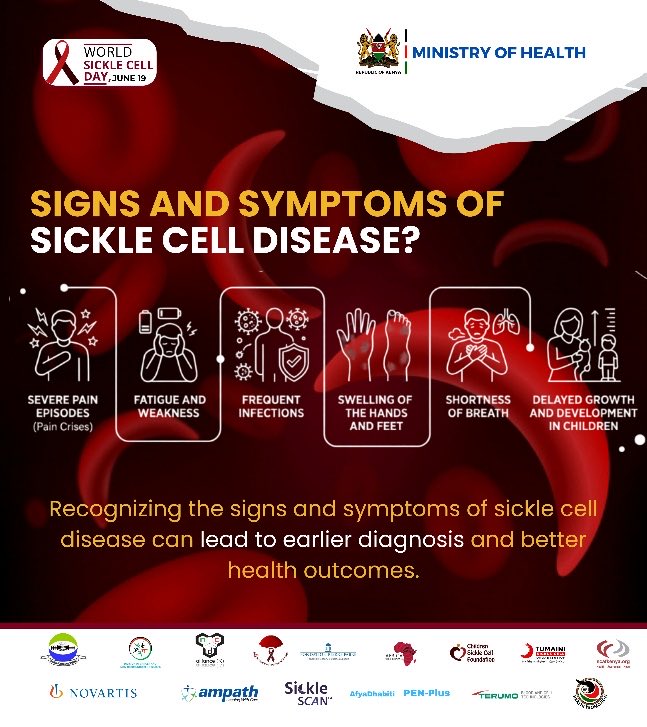
Symptoms of sickle cell disease typically appear in early childhood and vary significantly in severity from person to person. The most common clinical sign is the vaso-occlusive crisis, or pain crisis, which occurs when sickle-shaped cells block blood flow through tiny blood vessels. The National Heart, Lung, and Blood Institute (NHLBI) notes that these episodes can last from a few hours to several weeks and may affect the chest, abdomen, joints, and bones.
- Anemia: Chronic fatigue, shortness of breath, and pale skin resulting from the rapid breakdown of sickle cells.
- Dactylitis: Swelling and inflammation of the hands and feet, often one of the earliest symptoms in infants.
- Infections: Increased susceptibility to bacterial infections, such as pneumonia, due to splenic damage.
- Vision Problems: Retinal blood vessel blockage that can lead to permanent vision loss if left untreated.
- Delayed Growth: Slower physical development and delayed puberty in children and teenagers.
How Is Sickle Cell Disease Diagnosed?
Diagnosis is primarily achieved through blood testing. In the United States, almost all newborns are screened for SCD as part of state-mandated newborn screening programs, according to the American Society of Hematology (ASH). This early detection is critical for initiating preventative care, such as prophylactic antibiotics and vaccinations, which significantly reduce the risk of life-threatening infections in infancy.
For adults or older children who were not screened at birth, a hemoglobin electrophoresis or a high-performance liquid chromatography (HPLC) test can confirm the presence of abnormal hemoglobin. These tests identify the specific type of sickle cell trait or disease a patient carries, which is essential for both medical management and genetic counseling.
Current Approaches to Disease Management
While there is no universal cure for sickle cell disease, modern medical interventions have drastically improved life expectancy. The Mayo Clinic highlights several standard treatment pillars:
| Treatment Type | Purpose |
|---|---|
| Hydroxyurea | Reduces the frequency of pain crises and the need for blood transfusions. |
| Blood Transfusions | Increases the number of normal red blood cells to help prevent strokes and organ damage. |
| Gene Therapy | Newer, FDA-approved treatments that modify a patient’s own stem cells to produce healthy hemoglobin. |
| Stem Cell Transplant | The only current potential cure, typically reserved for severe cases in children with matched donors. |
Why Early Intervention Matters
Early medical intervention changes the trajectory of the disease. According to the NHLBI, children diagnosed through newborn screening are significantly more likely to reach adulthood than those diagnosed later. Ongoing care typically involves a multidisciplinary team, including hematologists, pain management specialists, and primary care physicians, to monitor for long-term complications like pulmonary hypertension and chronic kidney disease.
Patients are encouraged to maintain hydration, avoid extreme temperatures, and stay up to date on all vaccinations to prevent triggers for vaso-occlusive crises. As research continues to advance, particularly in the fields of gene editing and pharmacotherapy, the outlook for individuals living with sickle cell disease continues to improve.