{kind=link}
Multisystem Langerhans Cell Histiocytosis: Clinical Presentation and Diagnostic Challenges
Multisystem Langerhans Cell Histiocytosis (LCH) is a rare, complex disorder characterized by the abnormal proliferation and accumulation of Langerhans cells—a type of dendritic cell—that can infiltrate multiple organ systems. According to the Histiocytosis Association, the condition occurs when these cells, which normally help regulate the immune system, mutate and begin to damage healthy tissues. While LCH can manifest as a single-site bone lesion, multisystem involvement presents significant diagnostic challenges due to its ability to mimic more common pediatric infections or inflammatory diseases.
Understanding the Pathophysiology of LCH
At its core, LCH is now classified as a myeloid neoplasm. Research published in the Journal of Clinical Oncology confirms that over 50% of LCH cases involve a somatic mutation in the BRAF V600E gene. This genetic driver causes the uncontrolled growth of histiocytes, which then migrate to various sites, including the skin, liver, spleen, bone marrow, and central nervous system. Because these cells can infiltrate almost any organ, clinical symptoms are highly variable, often leading to delayed diagnosis in pediatric patients.
Common Clinical Manifestations
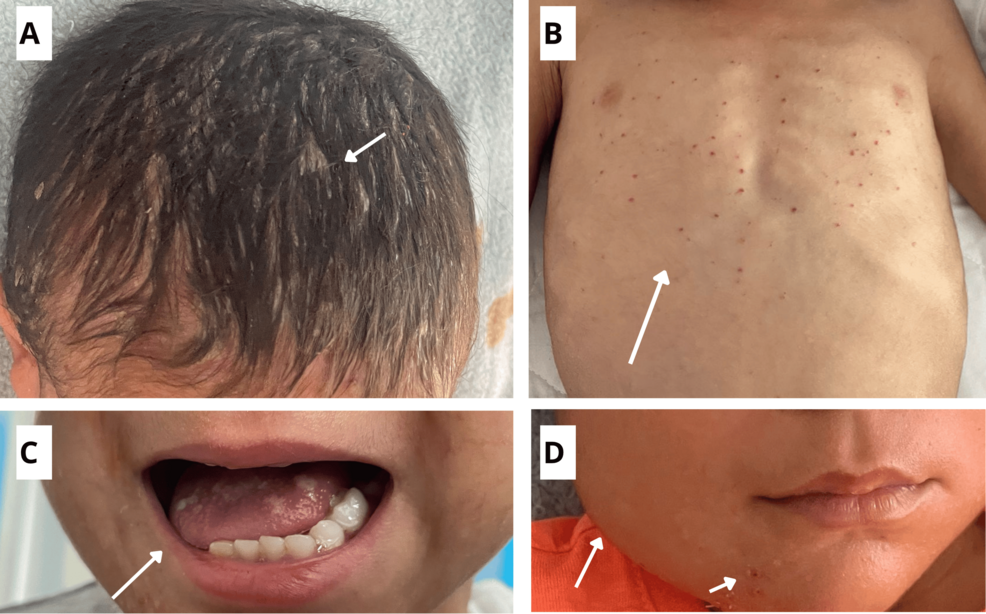
The presentation of multisystem LCH varies significantly depending on the organs affected. The National Cancer Institute identifies several hallmark symptoms that clinicians monitor:
- Dermatological: A persistent, scaly rash often mistaken for seborrheic dermatitis or diaper rash.
- Skeletal: Painful, lytic bone lesions, most commonly found in the skull, ribs, or long bones.
- Endocrine: Diabetes insipidus, caused by infiltration of the pituitary gland or hypothalamus.
- Systemic: Unexplained fevers, failure to thrive, hepatosplenomegaly (enlarged liver and spleen), and lymphadenopathy.
Diagnostic Approaches and Management
Diagnosing multisystem LCH requires a multidisciplinary approach. Because the disease is rare, initial symptoms are frequently attributed to common childhood ailments. According to the StatPearls medical database, the gold standard for diagnosis is a biopsy of an affected site, accompanied by immunohistochemical staining to confirm the presence of CD1a and Langerin (CD207) protein markers on the abnormal cells.
Once diagnosed, patients undergo a “skeletal survey” and systemic imaging, such as PET or CT scans, to determine the extent of organ involvement. Treatment strategies are risk-stratified based on whether the disease is “risk-organ positive” (involving the bone marrow, liver, or spleen) or “risk-organ negative.” Current standard-of-care protocols often involve systemic chemotherapy, such as vinblastine and corticosteroids, though targeted therapies—specifically BRAF inhibitors—are increasingly used in cases that are refractory to traditional treatment.
Key Takeaways for Clinical Awareness
| Feature | Clinical Detail |
|---|---|
| Primary Driver | BRAF V600E mutation in >50% of cases. |
| Diagnostic Marker | CD1a and CD207 positivity via biopsy. |
| High-Risk Sites | Liver, spleen, bone marrow, and central nervous system. |
| Common Misdiagnosis | Chronic skin rashes, osteomyelitis, or persistent ear infections. |
Future Directions in Research
The shift toward viewing LCH as an inflammatory myeloid neoplasm has changed how researchers approach treatment. By targeting the MAPK pathway, physicians are moving toward more personalized medicine. While the prognosis for multisystem LCH has improved significantly over the last two decades, long-term follow-up remains essential. Survivors are at risk for late effects, including neurodegenerative changes and secondary endocrine dysfunctions, necessitating lifelong surveillance by specialized oncology teams.
Related reading