{kind=link}
Viral DNA Load and Genetic Factors: New Insights from Large-Scale Biobank Studies
Recent research leveraging data from large biobanks like the UK Biobank and the NIH All of Us Research Program is shedding new light on the complex interplay between human genetics, viral infections, and health outcomes. A growing body of evidence suggests that genetic factors influence both the presence and abundance of various viruses within the human body, potentially impacting disease susceptibility and progression. This article explores the key findings from these studies, the methodologies employed, and the implications for future research.
Genome-Wide Association Studies and Biobank Collaboration
A collaborative initiative involving 24 biobanks across five continents, the Global Biobank Meta-analysis Initiative (GBMI), has enabled researchers to conduct more powerful and diverse genetic studies. This initiative has grown to include data from over 2.2 million people, facilitating the identification of genetic variants linked to disease and traits. Global Biobank Meta-analysis Initiative. The studies analyzed genetic, clinical, and other data to identify genetic variants associated with specific traits or diseases.
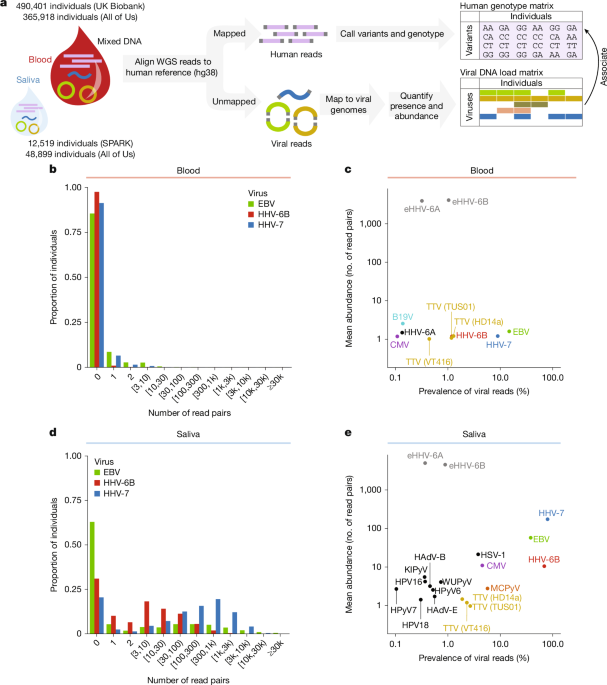
Researchers performed genome-wide association studies (GWAS) on data from the UK Biobank, the NIH All of Us (AoU) cohort, and the Simons Foundation Autism Research Initiative (SPARK) to profile viral DNA load. Whole genome sequencing (WGS) data was generated in previous studies, utilizing PCR-free methods and Illumina NovaSeq 6000 machines. Pan-UK Biobank genome-wide association analyses.
Key Findings: Viral DNA Load and Genetic Associations
The research identified significant associations between genetic factors and the presence and abundance of several viruses, including Epstein-Barr virus (EBV), human herpesvirus 6B (HHV-6B), and human herpesvirus 7 (HHV-7). Notably, the study found 14,676 significant loci (P < 5 x 10-8) in meta-analysis that were not found in the European ancestry group alone, highlighting the importance of including diverse populations in genetic research. Pan-UK Biobank genome-wide association analyses.
Specific findings include:
- Associations between the CAMK2D gene and triglyceride levels. Pan-UK Biobank genome-wide association analyses
- A known pleiotropic missense variant in G6PD associated with several biomarker traits. Pan-UK Biobank genome-wide association analyses
- Genetic influences on viral DNA load, particularly for EBV and HHV-7.
- Potential causal relationships between EBV DNA load and certain diseases, identified through Mendelian randomization.
Methodological Considerations
The researchers employed rigorous quality control measures and analytical frameworks. This included:
- Selection of a panel of 31 viruses based on previous prevalence studies.
- Realignment of unmapped reads to viral genomes using BWA-MEM.
- Filtering of alignment data to remove regions with excessive coverage suggestive of misalignments.
- Use of linear mixed models (BOLT-LMM) to account for relatedness and population structure.
- Mendelian randomization to assess causality between viral DNA load and disease phenotypes.
Implications and Future Directions
These findings have significant implications for understanding the role of viruses in human health and disease. The identification of genetic factors influencing viral DNA load could lead to the development of targeted interventions to prevent or treat viral infections. Further research is needed to validate these findings in larger and more diverse populations, and to explore the functional mechanisms underlying these genetic associations.
The study highlights the power of collaborative biobank initiatives to accelerate genetic discovery and improve human health. By combining data from multiple sources, researchers can overcome limitations of individual studies and gain a more comprehensive understanding of complex biological processes.
Frequently Asked Questions
What is a biobank?
A biobank is a repository of biological samples (e.g., blood, saliva, tissue) and associated data. These resources are used for research to improve our understanding of health and disease.
What is genome-wide association study (GWAS)?
GWAS is a research approach used to identify genetic variants associated with a particular trait or disease. It involves scanning the genomes of many people and looking for common variations that occur more frequently in people with the trait or disease than in people without it.
What is Mendelian randomization?
Mendelian randomization is a method used to infer causal relationships between an exposure (e.g., viral DNA load) and an outcome (e.g., disease) using genetic variants as instrumental variables.
Worth a look